摘要
胆红素是人体红细胞代谢的重要产物,其代谢过程的平衡与否直接关系到机体的健康状态。当血液中胆红素水平超过正常范围时,便会引发高胆红素血症,表现为皮肤、巩膜黄染等典型症状。相对于胆汁酸,胆红素毒性较弱,对人体影响相对较小,在人群流行病学研究发现,血清胆红素轻度升高人群的恶性肿瘤、心脑血管疾病等患病率显著降低,被称为“黄色荷尔蒙”。胆红素的代谢需经历产生、转运、入肝、生物转化、排泄及重吸收等多个环节,每个环节的精密协作保障了胆红素的有序代谢。当红细胞破坏过多导致的胆红素 “原料” 过剩,转运载体缺陷、肝细胞功能异常、胆道排泄受阻等环节的障碍,都可能引发高胆红素血症,其中高胆红素类别、程度、时间等维度是疾病诊治临床思维导入的重要线索。本文将系统梳理胆红素在人体中代谢的各个阶段,用系列病例来阐述各个环节出现问题引起高胆红素血症的机制、临床特征及诊疗要点,为深入理解胆红素的代谢机制及不同病因所致高胆红素血症的特点,对于疾病的早期识别病因、精准诊断和有效治疗提供帮助。
胆红素在体内的代谢过程
胆红素分为间接胆红素(也称游离胆红素)与直接胆红素(也称结合胆红素),前者为非水溶性(脂溶性),正常情况下不可直接从胆管及尿中排出,需经肝脏这个“生化工厂”的转化为水溶性的直接胆红素,才能从胆管或尿液中排出体外。血清中的胆红素约90%是由衰老的红细胞破裂而来,产生间接胆红素,其为一种低毒性的人体代谢产物,但不能直接经胆道或尿液排除体外,间接胆红素入血后将和白蛋白结合形成胆红素-白蛋白复合物,在肝窦处胆红素-白蛋白复合物分离成胆红素,经转运蛋白主动转运进入肝细胞中;间接胆红素进入肝细胞后通过葡萄糖醛酸转移酶发生生物转化,形成水溶性的直接胆红素后,大多数经毛细胆管转运蛋白从肝细胞排入毛细胆管,随着胆汁进入肠道,大部分形成黄色的粪便排出体外(大便为黄色原因),小部分胆红素在回盲部经“肠肝循环”回流入体循环,并随尿液排出,因此正常尿液颜色呈浅黄色。在整个胆红素代谢过程中,任一环节的失调均会导致血清胆红素的增高。
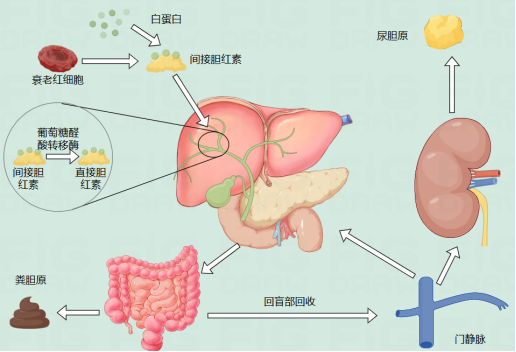
图一 胆红素代谢途径
胆红素代谢过程受损所致高胆红素血症
01 胆红素生成过多
病例一:蚕豆病致重度溶血性贫血一例
病例特点:患者男性,65 岁。因“皮肤巩膜黄染、尿黄 2 天”入院。入院前 2 天不明原因出现全身皮肤及巩膜黄染,小便呈浓茶样改变,伴头晕、乏力、口干、食欲减退。查血常规:红细胞 2.51 × 1012 /L,血红蛋白浓度 91 g /L(轻度贫血);肝功能:总胆红素 517.0 μmol /L,直接胆红素41.3 μmol/L,间接胆红素475.8 μmol/L(以间接胆红素升高为主),网织红细胞 8.31%,乳酸脱氢酶963 U/L。初步考虑为溶血所致总胆红素增高。追问患者发病过程,患者回忆发病前2日曾进食较多蚕豆,遂行葡萄糖6-磷酸脱氢酶( glucose-6-phosphate dehydrogenase deficiency,G6PD) 缺陷筛查,结果显示 G6PD 缺乏,故患者确诊为“G6PD 缺乏症所致溶血性贫血”。治疗上给予甲强龙静推( 40 mg / d,1 次/ d),营养对症支持治疗后 2 天复查肝功能:总胆红素 48.6 μmol /L,直接胆红素 11.8 μmol /L,间接胆红素 36.6 μmol /L;血常规:红细胞 2.74 × 1012 /L,血红蛋白浓度 95 g /L,尿常规恢复正常,遂停用甲强龙。患者经治疗后溶血情况得以改善,红细胞及血红蛋白水平缓慢回升,血清胆红素水平显著下降。
体内的胆红素80~90%由衰老的红细胞破坏所致,正常生理状态下,红细胞寿命约120天,衰老红细胞在单核-巨噬细胞系统中被破坏,血红蛋白分解为珠蛋白和血红素,血红素进一步转化为胆绿素(biliverdin),最终还原为间接胆红素。当各种原因导致红细胞破坏速度超过肝脏代谢速度时,胆红素的 “原料” 供应急剧增加,便会打破代谢平衡,引发以间接胆红素升高为主的高胆红素血症。 从病因角度,此类溶血可分为先天性与获得性两类。
先天性因素主要与红细胞自身缺陷相关,如遗传性球形红细胞增多症(红细胞膜结构异常)、葡萄糖- 6 -磷酸脱氢酶(G6PD)缺乏症(红细胞酶缺陷)、地中海贫血、卟啉病等。这些疾病因基因问题导致红细胞易被破坏,患者可出现慢性溶血,表现为持续性或间歇性黄疸,常伴随贫血、脾大等症状。获得性因素则包括自身免疫性溶血性贫血、新生儿溶血、非同型输血溶血、蚕豆病、蛇毒、阵发性睡眠性血红蛋白尿、药物等,其特点是起病较急,黄疸程度与溶血速度相关,严重时可出现急性肾衰竭等并发症。
临床特征上,溶血所致高胆红素血症以间接胆红素升高为主要标志(因肝脏对间接胆红素的摄取、结合能力达到饱和)。同时,患者多伴随溶血相关表现,如贫血(血红蛋白降低、红细胞计数减少)、网织红细胞升高(骨髓代偿性增生)、血清乳酸脱氢酶(LDH)升高(红细胞破坏释放)、游离血红蛋白升高或血红蛋白尿等。
02 间接胆红素转运障碍
白蛋白作为间接胆红素在血液中转运的核心载体,其数量与功能直接影响间接胆红素的代谢过程。正常情况下,间接胆红素(脂溶性)需与白蛋白结合形成 “白蛋白-间接胆红素复合物”,才能通过血液循环稳定运输至肝脏。当白蛋白缺乏或功能异常时,游离间接胆红素无法有效被携带,难以进入肝细胞代谢,进而导致血液中间接胆红素总量升高,形成高胆红素血症。从病因来看,白蛋白缺乏所致高胆红素血症可分为先天性与获得性两类。
新生儿出生前依赖母亲经胎盘脐带血管提供蛋白,出生后肝脏合成蛋白能力差,易引起新生儿黄疸,经紫外线照射后使皮肤表现的胆红素发生化学变化,使之成为更易被肝脏代谢或直接排出体外的形式,随着出生后肝脏合成功能逐步增强,黄疸也会逐步消失。先天性因素主要为遗传性白蛋白合成缺陷,此类疾病罕见,患者因肝细胞合成白蛋白的能力缺失,出生后即出现严重低白蛋白血症,间接胆红素无法正常结合转运,表现为持续性黄疸。获得性因素则更为常见,包括严重肝病(如失代偿期肝硬化、肝功能衰竭等)导致的白蛋白合成减少、肾病综合征等疾病引发的白蛋白丢失过多,或营养不良、慢性炎症等导致的合成原料不足。
03 间接胆红素在肝窦处入肝受阻
病例二:Rotor综合征一例
病例特点:患者女性,39岁,因“反复皮肤巩膜黄染37年,复发1周”入 院。患者于37年前(2岁时)无明显诱因出现皮肤巩膜黄染,无腹胀、腹痛、恶心、呕吐,无心慌、气急、纳差、乏力,无寒战、高热等不适,外院检查发现黄疸指数高,院外保肝治疗效果不佳,反复求治,先后就诊于多家医院均未明确病因,黄疸持续存在。近1周患者皮肤巩膜黄染加重,查血常规示:白细胞计数5.8×109/L、血红蛋白浓度121g/L;肝功:白蛋白38.1g/L、总胆红素83.2μmol /L、直接胆红素55.5μmol /L、间接胆红素27.7μmol /L、谷丙转氨酶15.6IU/L、谷草转氨酶15.4IU/L、γ谷氨酰转肽酶11.6IU/L;乙型肝炎血清标记物:乙型肝炎表面抗体阳性、乙型肝炎核心抗体阳性,甲、丙、戊 型 肝炎抗体及自身免疫性肝炎抗体均阴性;凝血功能四项正常。
肝穿活检示:肝小叶结构基本正常,肝细胞浊肿,窦周少许单个核细胞浸润,汇管区无界面炎症,未见纤维间隔形成(图二)。该患者肝脏组学基本正常,结合胆红素升高特点初步考虑为Rotor综合征。
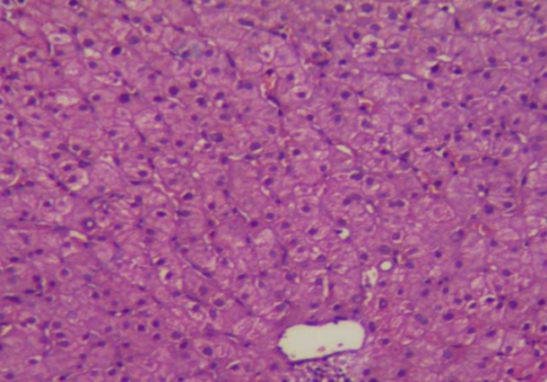
图二 肝穿病理:肝细胞间毛细胆管形态异常(扩张呈不规则池状萎缩、微绒毛变短、数量变少。
间接胆红素入肝转运蛋白障碍所致的高间接胆红素血症,是胆红素代谢异常中极具代表性的一类疾病,其核心机制与肝细胞窦状膜上的有机阴离子转运多肽(OATP)功能异常密切相关。其中,OATP1B1(由SLCO1B1基因编码)和 OATP1B3(由SLCO1B3基因编码)是摄取间接胆红素的核心转运体,二者通过特异性识别 “白蛋白-间接胆红素复合物”,经构象改变使间接胆红素从白蛋白解离并泵入肝细胞。当这些转运蛋白因基因缺陷、肝细胞损伤或抑制剂影响而功能受损时,间接胆红素无法正常进入肝细胞进行后续代谢,形成以间接胆红素升高为主的高胆红素血症。Rotor综合征为基因突变所致转运蛋白缺乏,同时合并毛细胆管排泄胆红素蛋白基因突变及功能降低,因此患者血清间接与直接胆红素均升高。获得性OATP1B1/1B3功能受损主要为药物所致(如环孢素、利福平、阿糖胞苷等),可引发药物性高胆红素血症,停药后黄疸多可缓解。
综上,间接胆红素入肝转运蛋白障碍所致的高胆红素血症,其本质是胆红素代谢通路中 “摄取环节” 的功能异常。深入理解这一机制,不仅有助于明确疾病诊断与鉴别,更为探索靶向修复转运蛋白功能的治疗策略提供了重要理论依据。
04 间接胆红素肝内生物转化受阻
肝细胞内的生物转化是间接胆红素代谢的核心环节,其核心过程为间接胆红素在肝细胞内与葡萄糖醛酸结合形成直接胆红素,这一反应主要由尿苷二磷酸葡萄糖醛酸转移酶(UGT)催化完成。当该环节因关键酶功能缺陷或缺失发生障碍时,间接胆红素无法正常转化为水溶性的直接胆红素,进而在血液中蓄积,引发以间接胆红素升高为主的高胆红素血症。从病因角度,肝细胞内生物转化障碍可分为先天性和获得性两类,均与关键酶的功能异常直接相关:
先天性关键酶缺失主要源于基因缺陷导致的 UGT 家族功能缺陷,其中最具代表性的是 Gilbert 综合征和 Crigler-Najjar 综合征。Gilbert综合征为UGT1A1 基因部分缺陷所致,患者表现为长期、不同程度间接胆红素升高,可在感染、疲劳或禁食后加重,而其他肝功能指标通常正常,这类患者可出现不同程度皮肤巩膜黄染,无特别治疗措施。Crigler-Najjar 综合征则为 UGT1A1 基因完全缺失或严重突变,患者出生后即出现严重黄疸:核黄疸,该类主要在新生儿科偶可见,如不能及时肝移植,患儿往往在出生后不久夭折。
获得性关键酶数质量降低由各种原因肝细胞损伤导致。常见病因包括病毒性、药物性、脂肪性、酒精性、自身免疫性、循环障碍、缺血缺氧、遗传代谢性、寄生虫及微生物感染、内分泌系统疾病及血液系统疾病等引起肝脏功能不同程度受损,使UGT数量减少、活性降低。与先天性疾病不同,获得性障碍常伴随转氨酶升高、白蛋白降低等肝细胞损伤证据,且黄疸程度与肝损伤程度平行,去除病因(如停用肝毒性药物、控制感染)后,酶活性可部分或完全恢复,黄疸随之缓解。




